
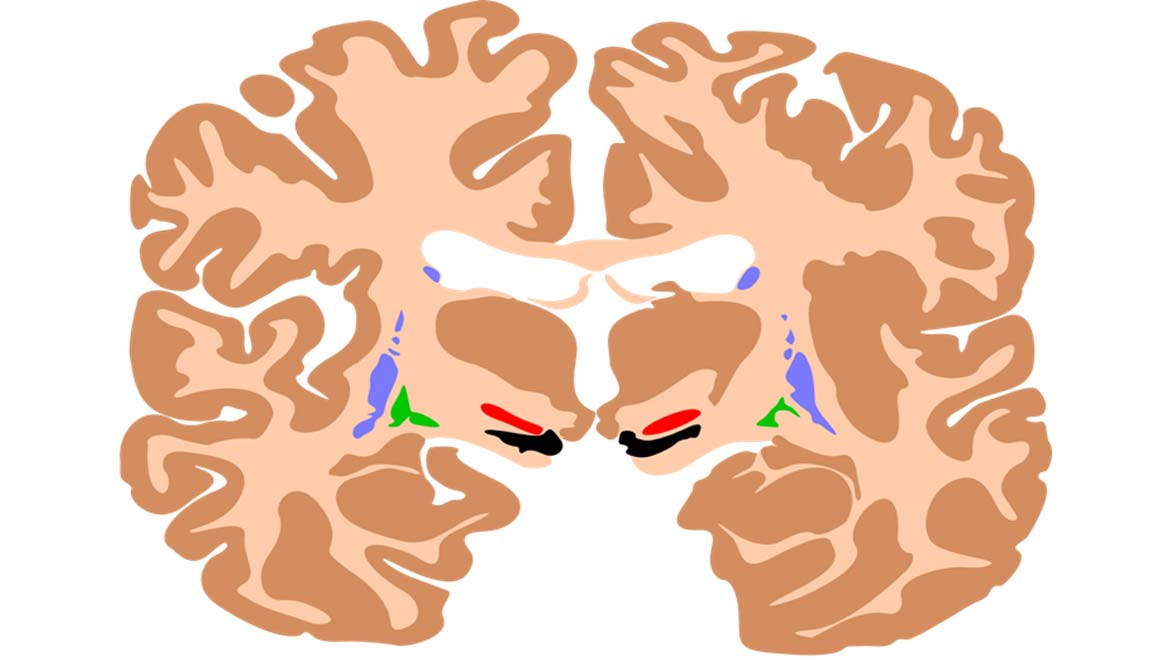
Parkinson's disease (PD) is a progressive neurological that is most strongly associated with motor and functional impairments. Its pathophysiology is currently thought to be driven by accumulations of the protein, alpha-synuclein, in certain regions of the brain. In this condition, patients may lose the ability to control their movements, over time.
Some ground-breaking studies also suggest that PD is an autoimmune condition.
A strong understanding of how PD develops and progresses is being matched with steady improvements in outcomes for many patients.
However, the research also suggests that there is still much we do not know about the disease, particularly its very early stages.
For example, a study recently published in the journal, Neurobiology of Aging offers evidence that PD may start off in a similar way to a separate category of conditions known as lysosomal storage disorders. These diseases lead to a possibly fatal accumulation of lipids in cells.
The Potential Link Between PD and Lipid Metabolism
The study assessed the failure to clear lipids, in the context of age, using an appropriate animal model.
The results included findings that the expression of an enzyme, that breaks down a lipid found in the brain, is reduced in older mice compared to younger mice. This enzyme, known as lysosomal glucocerebrosidase or GBA1, is coded for by the gene, GBA, a known risk factor for PD in humans.
Therefore, the researchers behind this study now posit that reduced GBA may be an unprecedentedly early warning sign of the condition in older adults. They also suggest that the gene may be used as a biomarker of PD risk in those without a hereditary risk of the disease.
PD and GBA
PD may be a genetic condition. But some patients develop the condition without a relevant familial medical history too. This is known as sporadic PD, and it is more difficult to predict or screen.
On the other hand, researchers have observed that patients who carry the genetic mutation associated with Gaucher's disease, which is also related to GBA inhibition, have a statistically greater chance of developing PD.
These individuals do not really develop Gaucher's disease but have one defective copy of the GBA gene. This condition predisposes them to a 7- to 10-fold increase in the risk of developing PD.
As Gaucher's itself is a lysosomal storage condition that leads to toxic accumulations of cellular lipids, it now seems as though PD may be on this spectrum.
To test this emerging hypothesis, a team of researchers from Oxford University and McLean Hospital (an affiliate institution of Harvard Medical School located in Belmont, Massachusetts) compared the glycosphingolipid (GSL) levels of older mice with those of younger mice.
GSLs are structural and functional cellular lipids that are found in the brain. They come in a variety of sub-types, such as glucosylceramide and glucosylsphingosine. GSLs are broken down by GBA1 in the lysosome (a specific zone within a cell where such molecules are synthesized). Some of them are also broken down outside the lysosome by GBA2, an isoform of GBA1. A few GSLs are precursors of gangliosides, which are necessary components of membranes of many cell types, including nerve cells.
Age vs. GSLs
The researchers compared the GBA-targeted GSLs in groups of mice that were 1.5 months old, and in other groups of mice aged 24 months.
They found that the glucosylceramide and glucosylsphingosine levels in the brains of older mice were significantly greater than in those of the younger mice.
The levels of glucosylceramide in the mice aged between 17 and 24 months were increased by nearly 164%, compared to those found in the mice aged between 1.5 and 3 months.
The numbers of some complex gangliosides (e.g., tetra-acetylneuraminated ganglioside 1b, di-acetylneuraminated ganglioside (DG) 1b and DG1a) were also significantly reduced in the brains of older mice compared to the younger rodents.
On the other hand, the researchers reported that the levels of lactosylceramide and monosialated ganglioside a1 (GM1a) were significantly reduced in the older mice compared to their younger counterparts. These molecules are GSLs that are broken down by other enzymes (e.g., beta-galactosidase).
Therefore, this study indicates that brain GSLs typically increase with age.
The scientists took these results to draw a conclusion: this process may contribute to the damage that PD-linked neurons (i.e., dopaminergic cells located in the substantia nigra (SN) of the midbrain) take at early stages in the condition.
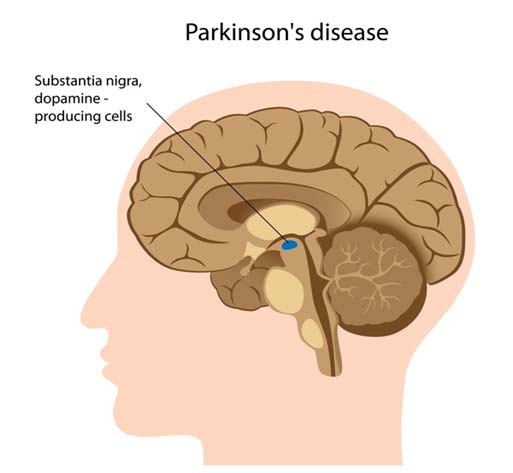
PD affects the substantia nigra in the brain, a region that controls balance and movement. (Source: NIH, Genetics Home Reference)
Furthermore, they may also explain previous observations that PD patients who lack mutations in GBA still exhibit reduced combined GBA1 and GBA2 activity. In other words, lipid build-up may occur with increased age, which, in turn, could influence the risk of PD development.
Overall, this study may not completely show concrete evidence that PD should be re-classified as a lysosomal disorder.
A 2015 study indicated that chronic GBA inhibition was associated with increased pathological alpha-synuclein in the SN of experimental mice. However, this paper did not suggest that the condition was associated with the inability to process some lipids and specific proteins in a normal fashion.
Such research also improves the case for GBA mutations as a risk factor for PD, which may lead to the enhanced ability to detect the disease at earlier stages, in the future.
References
P. J. Hallett, et al. (2018) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiology of Aging. 67: pp.189-200.
D. C. Schondorf, et al. (2014) iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 5: pp.4028.
E. Sidransky, et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 361:(17). pp.1651-1661.
E. M. Rocha, et al. (2015) Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis. 82: pp.495-503.
E. M. Rocha, et al. (2015) Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal. 23:(6). pp.550-564.
Top Image: Diagram showing some mid-brain regions, with the substantia nigra (implicated in PD) in black. (Source: Mikael Häggström and Andrew Gillies for Inkscape)



No comment