
Mitochondria are typically viewed as the ‘power station’ of the body. They are tiny cellular sub-units that supply cells with energy in the form of ATP. Mitochondria and their functions are so important that they have their own personal genome. This DNA is present in a singular ring (like that of a bacterium) and codes for essential mitochondrial proteins.
Scientific research had previously found that children receive this mitochondrial DNA (or mtDNA) only from their mothers. However, a new study recently published in the PNAS journal claims to have uncovered some rare cases in which paternal mtDNA is also present.
This breakthrough may lead to a considerable re-thinking of mitochondrial science and medicine.
Mitochondria are small, double-membraned organelles (or intercellular components) that are vital for the production of ATP, the biological units of energy. Besides this, mitochondria also function to generate iron-sulfur compounds in vivo (which are essential for certain protein and biomolecule types) and to balance cells' calcium levels. Therefore, if mitochondria are defective or fail, it can result in system-wide diseases for patients. In addition, many diseases, highly important or active tissues (e.g., the parathyroid, thyroid, eye, muscle, brain or various glands) are associated with mitochondrial dysfunction.
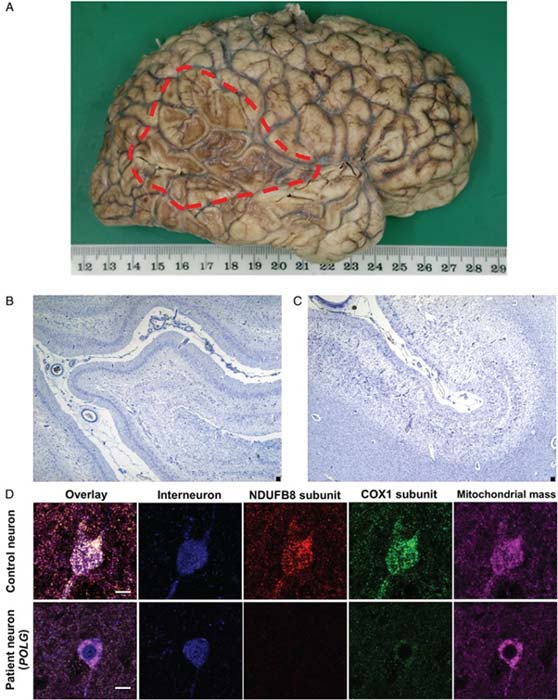
The brain tissue of a patient with a mitochondrial disorder (A), a cortical sample showing atrophy (the paler spaces; B % C) and cell-staining imaging studies with a normal control. (Source: C. L. Alston et al, 2017)
For example, the severe neurological condition, MELAS (mitochondrial encephalopathy, lactic acidosis and stroke‐like episodes) has been associated with mitochondrial failure. In addition, some forms of hypogonadism, deafness, retinopathy, and myopathy have been strongly linked to mtDNA mutations. This genome is a simple string of DNA connected at both ends to form a circle, as with bacterial plasmids, rather than in a helical complex like DNA. It contains 37 genes, which code for the precursors of mitochondrial proteins, the mitochondrial ribosome (which converts these precursors into proteins) and some parts of the oxidative-phosphorylation (OXPHOS) mechanism by which ATP is synthesized.
mtDNA is often present in multiple copies per cell; this number may vary from person to person and even from tissue to tissue. This situation, although possibly present to ensure mitochondrial density and normal healthy diversity (or homoplasmy), increases the risk of dangerous mutations (or heteroplasmy). This heteroplasmy may come in the form of point, insertion-deletion or deletion mutations within mtDNA. These mutations have been associated with specific conditions, including Kearns–Sayre syndrome (KSS), Pearson syndrome and MELAS.
For the vast majority of these mitochondrial diseases, no cure has been developed. However, mtDNA studies are contributing to an increasingly sophisticated understanding of the diseases that result from its mutation, and thus, possibly to improved therapies and options for affected patients in the future.
Mitochondrial disease research is further complicated by the fact that the ‘normal genome’ (or the one found in the nucleus of the cell that codes for the rest of it, and the rest of the biological information on the organism in question, for that matter) also contains about 1000 genes that determine the remainder of mitochondrial functions and structure. Mutations in these genes are also associated with the mitochondrial disorder, particularly as they make up most of the five complexes (I-V) of the OXPHOS mechanism.
Taken together, heteroplasmies and nuclear mitochondrial mutations account for diseases that affect at least 12.5 of every 100,000 adults and 4.7 of every 100,000 children.
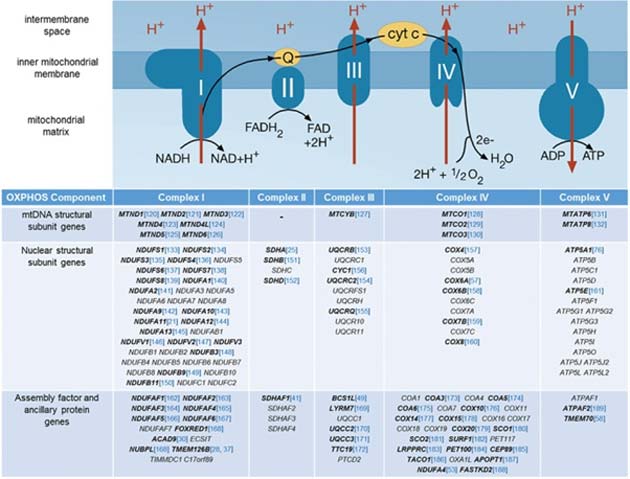
The OXPHOS mechanism, its complexes and the relative origins of their subunits. (Source: C. L. Alston et al, 2017)
75% of all mtDNA defects are inherited from parental sources. This source is thought to exclusively consist of the maternal gamete (or oocyte). The evidence that resulted in this generally-accepted conclusion has resulted in an overwhelming focus on oocytes in heteroplasmy research.
However, it now appears that scientists were somewhat misled in doing so. Dr. Shiyu Luo, a medical researcher at the Medical Center in Cincinnati’s Children's Hospital, may have kicked off a significant divergence in this science when he found a critical anomaly in a four-year-old patient with the symptoms of a mtDNA-related condition. 60% of the boy’s mtDNA matched that of his maternal grandfather. This meant that his mother had received the same amount of mitochondria from her father, and only 40% from her own mother as 'normal.'
Dr. Luo investigated further by analyzing samples of other families with a high level of heteroplasmy in the available medical records. This was done using whole-mtDNA sequencing and other methods at Cincinnati and at two additional College of American Pathologists-accredited labs.
The researcher and his team identified a total of 17 individuals from 3 discrete, unrelated families who had inherited mitochondria from both maternal and paternal sources. The existence of this unprecedented biparental mtDNA meant a sizeable upheaval and re-examination of the body of mitochondrial medical research completed to date.
On the other hand, this new form of mtDNA inheritance still appears to be extremely rare; the increased level of adaptation implied for the future of mitochondrial medicine may only apply to them.
All in all, it is fascinating, and somewhat fortunate for some individuals, to learn that even the most apparent rigid ‘rules’ of human biology, as we know them, can still be bent or even broken by discoveries such as this.
Top Image: An illustration showing a mitochondrion, its mtDNA and its size relative to a cell and its nucleus (background). (Source: By National Human Genome Research Institute (NHGRI) from Bethesda, MD, USA)
References
In Huge Shock, Mitochondrial DNA Can Be Inherited From Fathers, 2018, IFLScience, https://www.iflscience.com/health-and-medicine/in-huge-shock-mitochondrial-dna-can-be-inherited-from-fathers/, (accessed 28 Nov. 18)
C. L. Alston, et al. (2017), ‘The genetics and pathology of mitochondrial disease’, J Pathol, 241 (2), pp. 236-250
J. Chow, et al. (2017), ‘Mitochondrial disease and endocrine dysfunction’, Nat Rev Endocrinol, 13 (2), pp. 92-104
L. Craven, et al. (2017), ‘Recent Advances in Mitochondrial Disease’, Annu Rev Genomics Hum Genet, 18, pp. 257-275
S. DiMauro, et al. (2001), ‘Mitochondrial DNA mutations in human disease’, Am J Med Genet, 106 (1), pp. 18-26


No comment